Who we help > Biosimilar

Biosimilar
Introduction
Biosimilar medicines (or equivalent biological products) are not identical but should have no meaningful differences from the originator product in terms of quality, safety or efficacy Products of biotechnology are large and structurally complex proteins, which means that biosimilars cannot be developed in the same way as generic equivalents of small molecule originators. Developing a biosimilar requires a special skill set
The biosimilar market is soaring with approvals per year changing from single figures to double figures in 2017 in the EU. The increases are set to continue as both EU and US markets are benefitting from a range of factors such as high number of patent expirations of block busters, rising prevalence of chronic diseases and governments wanting cheaper medicines.
Number of approved biosimilars in the EU and the US, 2006 – 2018
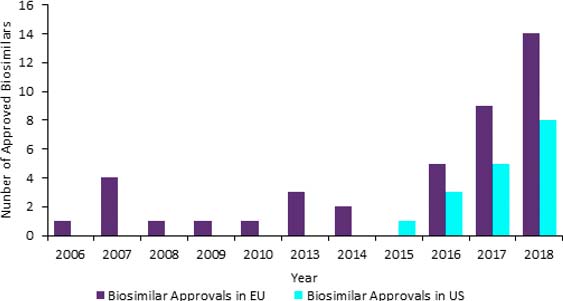
GlobalData: Drugs Database (accessed: 21 February 2019)
Are you developing a biosimilar?
Make sure that you have the regulatory resource and experience to get your candidate product to market ahead of the competition. Being the first biosimilar to market after patent and data exclusivity expiry of the innovator gives you a marketing advantage.
Not only do you need to pay particular attention to the regulatory requirements but you must also prepare to access your chosen market(s). The uptake of your new biosimilar is going to be dependent on you offering the product at a competitive price, with comparable safety and efficacy.
CambReg can help by:
- Providing honest unbiased regulatory advice
- Supplying information on latest relevant guidelines
- Provide you with a full understanding of the regulatory framework
- Prepare your NICE submission
- Recommend Experts in your field
Giving full consideration to all potential pitfalls at the start of your development programme usually means - early to market.
For economy and the best chance of long term success you should aim to satisfy Health Authorities in the EU, USA and Japan with one data package.
Development
We recommend seeking Scientific Advice preceding development of a biosimilar even if simply for reassurance that what you plan to do meets with the Health Authorities approval
Quality (Chemistry, Manufacture and Control - CMC)
It is well know that there will be CMC issues throughout the life of any product, the challenges are even greater with biosimilar products because of the complexity of their structure.
Let us help you to identify and avoid common pitfalls are such as:
- Poor identification of minor differences in structure (e.g. post-translational modifications)
- Flawed criteria used in selecting the Reference Product
- Production methods not being commercially scalable
- Lack of justification for impurity-profile differences
Our biotech knowledge and experience can help you decide on your CMC strategies before you embark on your clinical programme.
Take advantage of our strong in-house CMC team including Dr Mike James (ex-MHRA CTA assessor with more than 30 years' regulatory experience) and choose the most cost-effective route for your product
Our Services
- Gap analysis
- Scientific Advice
- GMP Compliance; Audits and Mock Inspections
- Recommendation of relevant Contract Houses
Non-Clinical
At this stage you will be required to carry out intensive in vitro testing generally coupled with studies in various animal species.
Questions that you should be asking at this stage:
Is one study enough to obtain the necessary PK, PD, toxicity and immunogenicity data?
Have you considered immunological properties to design your proof-of-concept studies?
Have you considered species specificity and other factors prior to your studies?
Do you need to assess toxicokinetics and recovery?
If you are unsure about any component of the non-clinical development, the solution is to outsource.
Our Services include:
- Review of and advice on your non-clinical programme
- Recommendation of contract houses for your pre-clinical studies
- Recommendation of opinion leaders in your field
- Arranging GLP Compliance Mock Inspections
Using a pragmatic approach and good arguments, we could help you satisfy the regulators with a less costly and streamlined non-clinical programme.
Clinical
In this most costly phase, make sure that you are working with experts.
Questions that you should be asking at this stage:
Can you substitute confirmatory clinical trials with pharmacodynamic studies?
Have you considered obtaining Scientific Advice from the regulators for your immunogenicity issues?
Have you fully defined and justified your a priori equivalence margin?
Our Services:
- Clinical programme advice
- CTA (Writing and Submission)
- Ethics Committee Approval
- Recommending opinion leaders
- Recommending a CRO to execute your clinical trial programme
- Mock GCP Inspections
Take advantage of our knowledge and experience during this important phase of your product development
Gaining Approval
Dossier Compilation and Procedure Management
Plan for early submission by building an eCTD dossier as you move through your development programme, this can save up to 6 months.
For submissions in the EU you will need to use the Centralised Procedure. Dossier preparation and dialogue with the European Medicines Agency (EMA) should ideally commence at least 12 months before the intended submission date.
Our Services:
- Preparation of all CTD Modules (1-5)
- Writing EU Risk Management Plans (RMP)
- Writing Environmental Risk Assessments (ERA)
- Preparation of Product Information (labels, leaflets and SPC)
- User Testing of Package Leaflets
- eCTD publishing
- Vaccine Antigen Master File Applications
- Plasma Master File Applications
- Management of the procedure from Pre-Submission to Post-Approval Phase Commitments
- Pre-procedural meeting
- Pre-submission requests
- Advice on Product Name
- Eligibility requests for Accelerated / Conditional Approvals
- Advice on fee waivers / reductions
- Preparation for Pre-Approval Inspections (GMP / GCP)
Post Approval
Our Services:
We can manage your MA portfolio and provide:
- Optimum variation submission strategies
- Advice on Sunset Clause Legislation
- Annual Safety Reports (ASR)
- Periodic Safety Update Reports (PSUR)
- Renewals
- Change of Ownership
- Pricing and Reimbursement
- Vetting of Advertising Materials
- Guidance on submission of post-approval data (including paediatric)
- QA system for dossier maintenance (Registered Technical Details (RTD))
- Pharmacovigilance and Medical Information services
We at CambReg have put together a unique cost effective post-licencing ‘package’ that can be adapted to suit your post-licencing needs whatever they are. We run a QA system for dossier maintenance using our in-house developed Registered Technical Details (RTD) template.
Opportunities
Take full advantage of your very valuable asset!
Post-licencing need not be a non-profit generating burden.
We would like to advise you on post-licencing activities that will allow you to exploit your products to their full potential such as:
- Prepare for a new indication or line extension
- Increase the number of ex EU markets with a Certificate of Pharmaceutical Product (CPP)
- Investigate the possibility of ‘biosuperiors’
- Push the boundaries with your advertising material
No EU regulatory expert in-house?
We can maintain your MA portfolio until you have reached a size where it becomes essential to have your own in-house regulatory staff.
Licensing in or out?
Due diligence is one of our specialties.